

Journal of Creation 38(2):62–67, August 2024
Browse our latest digital issue Subscribe
Enantiomeric amplification of amino acids: part 9—enantiomeric separation via crystallization
For some amino acids (AAs), DL racemic crystals are more soluble than pure D or L crystals, and vice versa for other AAs. Experiments to amplify small initial excesses of L enantiomers are critiqued here for lacking realism under natural conditions:
- Excessively high concentrations of pure AAs would have been required.
- Solution temperature would have to be kept fixed as water slowly evaporated.
- At just the right time the two phases would need to have separated whereas evaporation would more likely have completely desiccated the mixtures.
- Enriched AAs would have redissolved after rainfall, tidal incursion, or other sources of water in addition to mixing with racemic AAs from the environment.
- L-AAs solidified into crystals would have been unable to participate in the putative prebiotic origin of life chemistry.
Experiments wherein depleted D-enantiomers in a solution phase were replenished by adding complex catalysts to enhance racemization overlook that such catalysts would have racemized all kinds of AA indiscriminately. Rapid stirring while continually grinding crystals with glass beads does not reflect natural processes either.
We continue here, in part 9, a series of papers that evaluate the main proposals from the Origin of Life (OoL) community on how a small enantiomeric excess (e.e.) of D- or L-amino acids (AA) could have been naturally amplified. All the proposals have been found to be irrelevant or inadequate to explain how proteins based on only L enantiomers could have arisen through natural processes.
Amino acid conglomerates vs racemic compounds
When both enantiomers of an amino acid (AA) are present in solution, crystallization produces either DL enantiomer mixtures within each crystal or the crystals are composed of only D and L enantiomers. When the mixed enantiomers are combined 1:1 within the same crystal, the result is called a racemic compound (e.g., see figure 1, right panel). Alternatively, mixtures of individually pure D and L crystals are known as conglomerates (figure 1, left panel). Pasteur was able to separate D from L crystals of tartaric acid using tweezers and a microscope.1
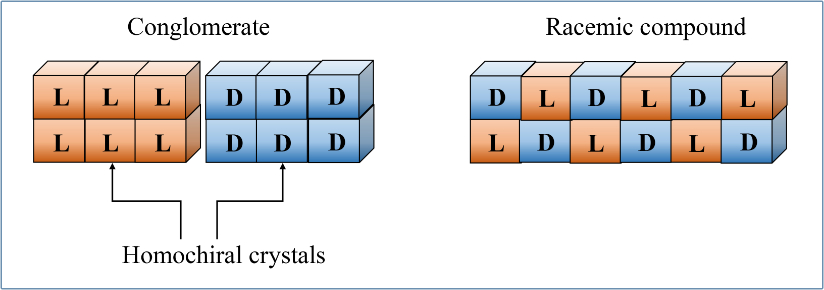
The difference in the two types of crystals is determined by whether the stronger interaction in the solid phase is between heterochiral (racemic) or homochiral (conglomerate) enantiomers. Racemic compounds are formed by ~90% of all known chiral compounds, and around 17 of the proteinogenic amino acids,2 although in many of the AA cases the ΔG is <2 kJ/mole.3
Physical separation of AA enantiomers is possible if they form racemic crystals (DL) having different solubility than their enantiopure compounds.4,5 This occurs when the free energy of formation for the mixed DL crystal is sufficiently different than for the pure crystals.6 Solubility differences could be affected by temperature, pH, ionic strength, or other factors.
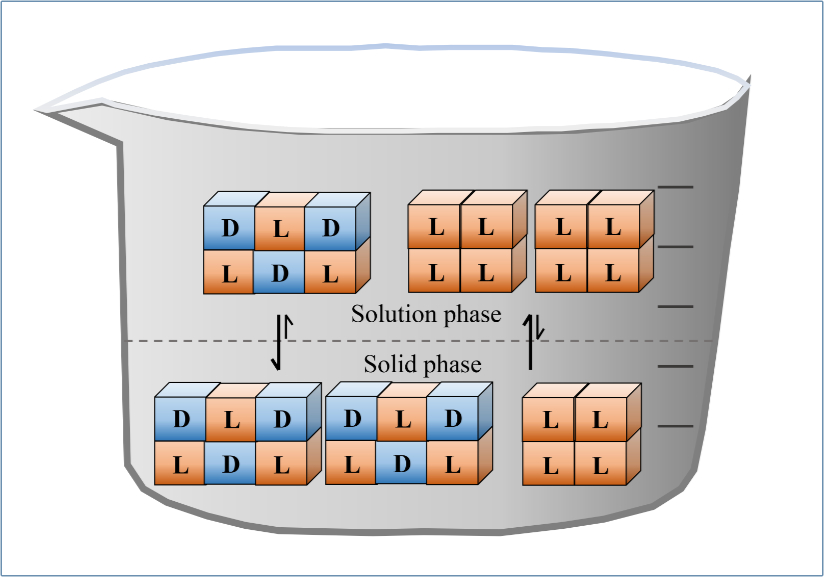
Enantiomeric enrichment using the liquid phase
Consider the case in which the racemic compound of an AA is less soluble than its homochiral crystals. Suppose that solutions are prepared with one enantiomer in excess. As the racemic material preferentially crystallizes, the e.e. (of the more soluble isomer) would increase in the solution phase, see figure 2.2 This separation can occur as the temperature is lowered or the concentration increased through evaporation. This provides the potential to separate solvent which is enriched with one enantiomer from the racemic solid phase.
Amplification of phenylalanine and tryptophan
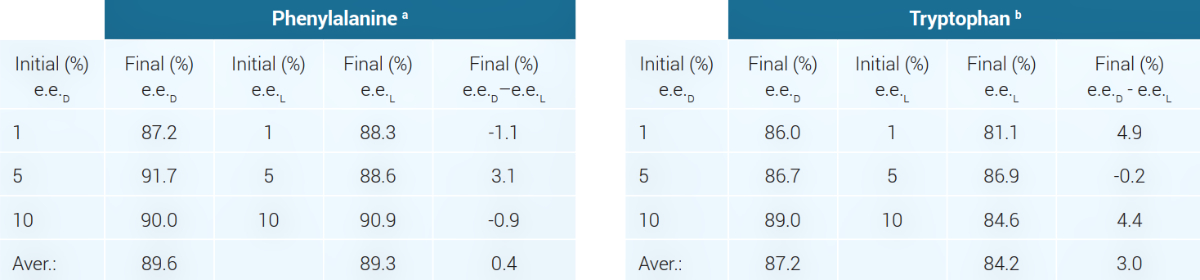
For most AAs found in proteins, the racemic crystals are less soluble than crystals composed of a single enantiomer. Breslow and Levine reported in 2006 that they prepared an aqueous solution containing ~500 mg of phenylalanine with a 1% excess of the L component and allowed it to slowly evaporate until the majority of the material (>400 mg) had crystallized.7 Surprisingly, the experimental details were not provided, such as temperature, the volume of water, duration or rate of evaporation, and whether stirring was used. The crystals were racemic, and the solution now contained phenylalanine having a 40% e.e.L. They then prepared a new aqueous solution with 500 mg of a 40% L e.e. and again slowly evaporated the water. Racemic crystals (racemate) again crystallized and were again separated. The supernatant solution now contained ~100 mg of phenylalanine having a ~90% e.e.L. This represented an L/D ratio of 95/5.8 The mirror results were obtained when they began with an excess of D-phenylalanine.6
Experiments were also performed beginning with aqueous solutions having 1%, 5%, or 10% e.e., which also led to an e.e. of up to 90% for phenylalanine. Similar experiments were conducted using tryptophan,9 and the results from both papers are summarized in table 1.
The scenario resembles a process carefully designed by chemical engineers, and would not have occurred without expert guidance for the reasons discussed next.
The use of pure AA solutions
Prebiotically very rare phenylalanine and tryptophan would have been present in ppb concentrations, based on thermodynamic, Miller-type experiments, and meteorite composition studies.10-12 Concentrated aqueous solutions could not have existed since contaminants (such as NaCl from seawater) would have been present in concentrations many orders of magnitude greater. Realistically, evaporation would have produced a dense slurry of chemicals containing virtually none of these AA.
Ideally controlled crystal separation conditions
Slow evaporation under carefully controlled conditions was used. Under natural conditions there would have been temperature fluctuations; e.g., between day and night or seasons, forming and dissolving LL, DD, and DL crystals. At any time, it would have been very rare for a solution to have contained predominantly pure DL-precipitated crystals.
Perfect timing for crystal separation
The researchers watched until >80% of the AA had crystallized and then measured the e.e.L of the liquid. Levine et al. speculated that
“Amplification via evaporation of water could have occurred on prebiotic earth in a drying lake bed near a site of meteorite landing. Preferential dissolution may have occurred when river or rainwater passed over an amino acid mixture, dissolving the single enantiomer with enriched enantiopurity and carrying it downstream.”9
Creating a measurable e.e.L from chemicals provided from a meteorite is far fetched. The lake would have initially contained racemic AA, and it is not plausible that meteorites would have provided the necessary e.e.L.13 Furthermore, the proportion of AA to other chemicals from a meteorite would have been negligible. In any event, the authors visualized rivers and rainwater waiting until much DL crystallization occurred and then extracting some of the supernatant, instead of diluting the lake water and redissolving the crystals, as would have occurred naturally. This is further discussed under ‘Optimization of all details’.
Remixing prevented
The fortuitous separation of racemic crystals and L-enriched liquid is contrived. Rainfall or extraneous water incursions would have simply remixed everything. For example, with temperature changes and addition of water, the DL crystals in the lake would have redissolved and mixed with previously separated L-enriched water. In addition, homogenous L crystals evaporated near the shore from L-enriched water would have easily redissolved back into the lake.
Evolutionary dead end
LL crystals precipitated from highly L-enriched water would not have served any purpose for OoL models.
Optimization of all details was necessary
The scenario requires e.e.L to increase faster than would be lost through racemization and mixing with racemic AA. However, what might a realistic scenario resemble? Consider a lake containing a miniscule proportion of dissolved phenylalanine with 1% e.e.L, mixed with many other chemicals. It is subject to random rainfall or contact with rivers, evaporation, and temperature fluctuations. Making the questionable assumption that some DL crystals did precipitate (despite the insignificant concentration of AA), what might the e.e.L of the separating L-enriched water be? The extracted water would have been diluted and contaminated with racemic AA by the new water transporting it, so >1% is not reasonable. L-enriched water could have desiccated near the lake, but what would have prevented rainfall from remixing everything later? Importantly, note that the excess L enantiomer being slowly removed would have automatically decreased the amount of excess L remaining behind in the lake.
Of course, water extracted from a drying lake after a considerable amount of LL crystals had also precipitated would have been even less enriched after mixing.
Nevertheless, assume some L-enantiomer–enriched water had separated. It would need to then undergo multiple additional cycles of amplification. However, the amount of AA now present would decrease with each cycle since, by assumption, crystals of DL had been removed. The resulting low concentration of AA would soon no longer be able to form crystals. Separating enriched water by first adding water and then evaporating it would also have continually increased the proportion of contaminants.
The researchers were only able to obtain e.e.L incrementally because every cycle was initiated with a pure sample of 500 mg AA having about the maximum theoretical e.e.L achievable during the preceding cycle.7
It is interesting that OoL researchers draw much attention to cases of e.e.L which are 1% or less, whether from meteorite samples or laboratory experiments. There is a high probability that these could be laboratory artifacts. But according to the duplicate experiments summarized in table 1, an average e.e.D of 87.2% was found for tryptophan, but only an average e.e.L of 84.2% for the L enantiomer. This is a considerable difference and in the wrong direction, since an excess of the L form is needed.
Suppose the goal of an experiment had been to show that an e.e.D could have arisen naturally (instead of the L form). It is very likely that this 3% average net difference, based on repeated experiments, would have been presented as supporting the wished-for hypothesis. Anomalous results are often interpreted as flawed and unlikely to be followed up on than results congruent with a favoured theory. This behaviour leads to a statistical distortion whereby more evidence will be collected to support the researcher’s view than to potentially refute it.14
Enantiomer enrichment using the solid phase
We will now switch to the case in which the racemic compound of an AA is more soluble than its homochiral crystals. In this case, beginning with an excess of one enantiomer in solution could produce homochiral crystals under the right conditions.2 This would lead to an e.e. in the solid phase.
Among the proteinogenic AAs, racemic asparagine (Asn) and threonine (Thr) can crystallize from aqueous solutions as conglomerates under the right conditions.15,16 Albrecht claimed, in 1943, that methionine also forms crystalline conglomerates, but this does not seem consistent with a detailed eutectic study, published in 2009 by Polenske, for temperatures between 1–60°C.17,18
For preferential crystallization to work for conglomerating crystals, the homochiral interactions must be stronger than heterochiral interactions at the crystal–aqueous interface. Since the solubilities of both enantiomers are identical, saturated solutions will contain an equal number of D and L molecules regardless of how great the overall difference between the amount of D and L in the conglomerate. This results in the potential for enantioenrichment in the solid phase.2
Critique of these studies
The same critiques apply here as above for those racemic compounds which were less soluble than the homochiral crystals; for example:
- For some AAs, enrichment required processing the solution phase, and for others the solid phase. A single natural process does not apply to all biological AAs.
- Nature would not have conveniently evaporated enough water until the L enantiomer in excess began to solidify and then quickly separated the enriched (in D enantiomer) liquid phase. Realistically, if enough water had evaporated to achieve a high concentration of the L crystals, then a little more evaporation would have caused the pure D and DL crystals to also crystallize from solution.
- The initial AAs would have been mixed with many other substances—instead of being highly pure and concentrated.
- Upon removing the L enantiomer, the solution phase would have become depleted in L compared to nearby water and would have decreased its e.e.L.
- One heavy rainfall or tide would have dissolved all the L-enriched crystals, racemizing the solution.
- Crystals containing a high e.e.L would have served no purpose for OoL purposes unless they first redissolved in water, interacted with other AAs, and formed peptides. But as soon as they dissolved, they would have begun to racemize and been contaminated with water less enriched (if at all) in L enantiomer.
Enrichment by forming larger pure D or L crystals
Under special laboratory conditions, some AAs can be enriched if one enantiomer is present in sufficient excess, permitting it to form larger crystals than the other enantiomer would. This occurs because the larger pure D or L crystals are less soluble than smaller ones. The solution phase will eventually become depleted in the formerly major enantiomer. However, this would be counteracted if the ‘wrong’ enantiomer had enough time to racemize in the solution phase.
For this enrichment process to work, the larger homochiral crystals must attract more of the same enantiomer faster than DL crystals form, and the DL crystals must be more soluble than the homochiral ones.19-22 The left- and right-handed crystals will grow and dissolve at the same rate unless there is a greater amount of one chiral form since secondary nucleation can amplify the difference.23
In 2008 Noorduin et al. showed that a crystalline enantiomer of a chiral non-AA molecule could be produced, starting from a nearly racemic mixture using attrition enhancement.19 This was accomplished using the imine of 2-methyl-benzaldehyde and phenylglycinamide shown as ‘1’ in figure 3. This molecule exists as two enantiomers that form separate R and S solid-phase crystals (i.e., a conglomerate). It also racemized rapidly in methanol or acetonitrile solution phase in the presence of the special organic-base DBU (1,8-diazabicyclo[5.4.0]undec-7-ene).19
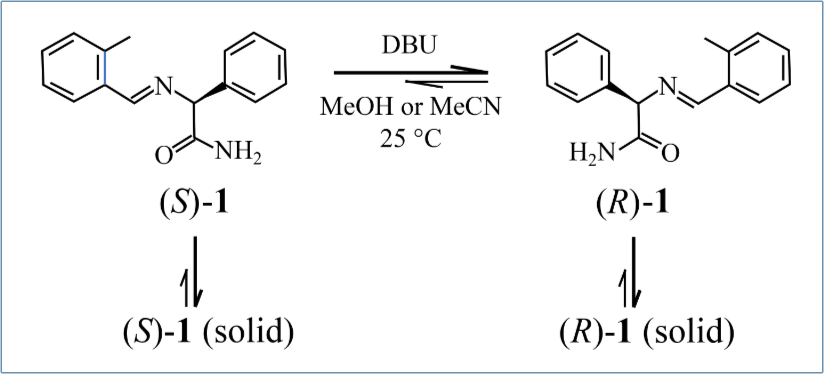
The experiments were initiated with solution-solid mixtures of (RS)-1 with various e.e. of (R)-1 or (S)-1 and magnetically stirred (1,250 rpm) at ambient temperature in the presence of 2.5 mm glass beads. Crystals of the majority enantiomer began to form first. This led automatically to enrichment in the solution phase of the other enantiomer.19 After solution-solid equilibrium was reached, the researchers forced solution-phase racemization by adding DBU as a catalyst, thereby replenishing the enantiomer lost through crystallization. With the help of the glass beads, which provided mechanical energy and continuous attrition of the crystals, the e.e. of the solid phase increased over time. This eventually led to a single enantiomer in the solid phase.19
The process seemed to rely on the fact that crystals having larger sizes grow faster than smaller crystals (called Ostwald ripening). This was initiated by the small e.e. of one of the enantiomers under continual attrition caused by agitation in the presence of glass beads.
Once enough of the solid crystals of a single enantiomer had formed, the system was ‘committed’, since competing nucleation to form crystals of the opposite enantiomer would be very difficult in the racemizing solution.19
Critique of these studies
- Large homochiral crystals based on only an L AA would need to grow rapidly and crystallize accompanied by the very rapid conversion of resulting excess D → L. Biogenetic AAs don’t have these properties. For most AAs, racemizing most of a ~1% excess of D under cold, crystallizing temperatures would have required hundreds of thousands or millions of years.24 During this time, any homochiral crystals already produced would have had countless opportunities to redissolve in rainwater or tides and to mix with racemic AAs.
- The racemization experiments did not involve AAs but molecules in special laboratory solvents like methanol or acetonitrile which are irrelevant for OoL purposes.
- Special organic bases like 1,8-diazabicyclo[5.4.0]undec-7-ene are irrelevant for OoL purposes. Racemizing catalysts on a primordial Earth would have been devastating since the last thing OoL scientists would want are substances able to racemize all AAs. The opposite is needed, that is a means to generate only L enantiomers for 19 biogenetic AAs. (Glycine does not possess a chiral carbon and therefore cannot exist as separate D and L enantiomers).
- The initial conditions required not just an e.e.L to permit the enrichment process to initiate, but a racemizing base, which was added at just the right time. Why would such a racemizing catalyst (which was soluble in the solvent used) not have already eliminated the initial e.e.?
- The necessary conditions have no natural analogue: rapid stirring at ~1,250 rpm, super-concentrated solutions of AAs, the presence of 2.5 mm glass beads for attrition, and a controlled ambient temperature.
- Random temperature changes, such as day and night transitions could have caused the entire AA content to freeze and contaminate any large homochiral crystals.
Deracemization by crushing crystals through mechanical stirring
Viedma performed studies beginning with a supersaturated aqueous solution of 50:50 L and D NaClO3 chiral crystals, stirred in the presence of glass balls with 3 mm diameter, usually stirred at 600 rpm using a magnetic bar.25 Sodium chlorate’s ions Na+ and ClO3- are not chiral, but the crystals they form are, typically produced in equal amounts of left- and right-handed crystals. The very first crystal formed could be based on either enantiomer.2
Under rapid stirring, this ‘Eve’ crystal was broken into thousands of smaller crystals by impact with the stirring bar. All the ‘daughter’ crystals had the same chiral form and grew by secondary nucleation which attracted ions from solution. If secondary nucleation occurred fast enough, a single chiral solid state was obtained, as shown in figure 4.2
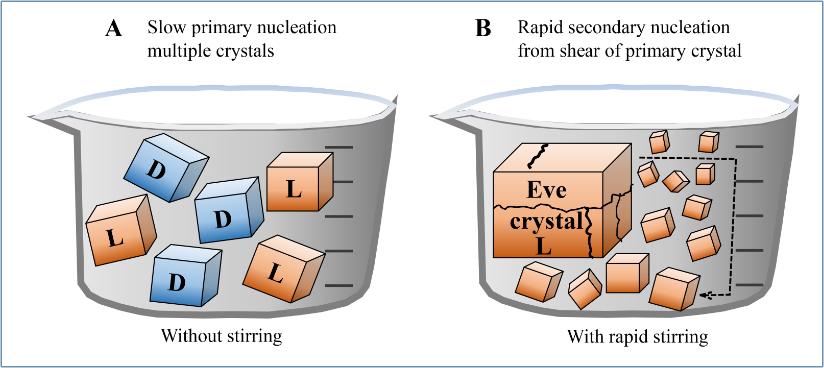
This effect was believed to be due to so-called Ostwald ripening, which was highly enhanced by the continuous abrasion-grinding process.20,26
Crushed between the glass balls, crystals were repeatedly broken down while others formed. After grinding for several hours, one chiral form of crystal predominated. Viedma reported that the dominant form which crystallized was random when the original mixture was racemic.20
In the absence of crystal attrition by glass balls, no excess of crystals was obtained, even with rapid stirring. Tests demonstrated that enantiomeric crystal formation depended on the concentration of the glass balls and rate of stirring.20
Viedma showed, in 2008, that under special conditions, this effect could be set up for one proteinogenic amino acid, namely aspartic acid.15
Critique of these studies
- Importantly, the experiments using NaClO3 produced D or L crystals with equal probability. If such processes could have occurred naturally, no e.e.L would have resulted.
- The unusual conditions necessary produced a laboratory artifact not expected to occur in nature. Rapid stirring was not enough; crystal attrition by glass balls was also necessary.
- Only the AA aspartic acid has been shown to produce these conglomerates.
References and notes
- Kauffman, G.B. and Myers, R.D., Pasteur’s resolution of racemic acid: a sesquicentennial retrospect and a new translation, The Chemical Educator 3(6):1–18, 1998. Return to text.
- Blackmond, D.G., The origin of biological homochirality, Cold Spring Harb Perspectives in Biology 11(3):a032540, 2019. Return to text.
- Huang, J. and Yu, L., Effect of molecular chirality on racemate stability: α-amino acids with nonpolar r groups, J. American Chemical Society 128(6):1873–1878, 2006. Return to text.
- Klussmann, M., Iwamura, H., Mathew, S.P., Wells, D.H. Jr, Pandya, U., Armstrong, A., and Blackmond, D.G., Thermodynamic control of asymmetric amplification in amino acid catalysis, Nature 441:621–623, 2006. Return to text.
- Klussmann, M, White, A.J.P., Armstrong, A., and Blackmond, D.G., Rationalization and prediction of solution enantiomeric excess in ternary phase systems, Angewandte Chemie, International Edition 47:7985–7989, 2006. Return to text.
- Morowitz, H.J., A mechanism for the amplification of fluctuations in racemic mixtures, J. Theoretical Biology 25:491–494, 1969. Return to text.
- Breslow, R. and Levine, M., Amplification of enantiomeric concentrations under credible prebiotic conditions, PNAS 103:12979–12980, 2006. Return to text.
- e.e.L = (L – D)/ (L + D) = 0.9; replacing D by= (1 – L) leads to 2L – 1 = 0.9. Therefore, L = 0.95 and D = 0.05. Return to text.
- Levine, M., Kenesky, C.S., Mazori, D., and Breslow, R., Enantioselective synthesis and enantiomeric amplification of amino acids under prebiotic conditions, Organic Letters 10:2433–2436, 2008. Return to text.
- Higgs, P.G., and Pudritz, R.E., A thermodynamic basis for prebiotic amino acid synthesis and the nature of the first genetic code, Astrobiology 9(5):483–490, 2009. Return to text.
- Parker, E.T., Cleaves, H.J., Dworkin, J.P., Glavin, D.P., Callahan, M., Aubrey, A., Lazcano, A., and Bada, J.L., Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment, PNAS 108:5526–5531, 2011. Return to text.
- Burton, A.S., Stern, J.C., Elsila, J.E., Glavin, D.P., and Dworkin, J.P., Understanding prebiotic chemistry through the analysis of extraterrestrial amino acids and nucleobases in meteorites, Chemical Society Reviews 41:5459–5472, 2012. Return to text.
- Breslow and Levine reported that reactions using L-α-methylvaline (presumably from a meteorite) produced the wrong D-phenylalanine in their chemical transformations!7 To revert this disastrous outcome, the reaction conditions (temperature, duration, etc.) were optimized, using ideal 4 equiv of 100% L-α-methylvaline (presumably from a meteorite) mixed with 1 equiv of sodium phenylpyruvate (of unknown origin) and 1 equiv cupric sulfate (of unknown origin).9 Such mixtures have no prebiotic plausibility. A maximum of 37% e.e. L-phenylalanine was obtained, based on 100% L-α-methylvaline. Of course, pure L-α-methylvaline would not have been available, and decreasing the proportion of cupric sulfate would have led to the incorrect D-phenylalanine. Return to text.
- Truman, R., Clean-up and analysis of small data sets can distort conclusions, J. Creation 36(2):66–71, 2022. Return to text.
- Bernal, I., Conglomerate crystallization and chiral discrimination phenomena: in vivo and in vitro, J. Chemical Education 69:468–469, 1992. Return to text.
- Tarasevych, A.V., Sorochinsky, A.E., Kukhar, V.P. and Guillemin, J-C., deracemization of amino acids by partial sublimation and via homochiral self-organization, Origins of Life and Evolution of Biospheres 43:129–135, 2013. Return to text.
- Albrecht, G., Schnakenberg, G.W., Dunn, M.S., and McCullough, J.D., Quantitative investigations of amino acids and peptides. xii. structural characteristics of some amino acids, J. Physical Chemistry 47:24, 1943. Return to text.
- Polenske, P. and Lorenz, H., Solubility and metastable zone width of the methionine enantiomers and their mixtures in water, J. Chemical Engineering Data 54:2277–2280, 2009. Return to text.
- Noorduin, W. L., Izumi, T., Millemaggi, A., Leeman, M., Meekes, H., Van Enckevort, W.J., Kellogg, R.M., Kaptein, B., Vlieg, E., and Blackmond, D.G., Emergence of a single solid chiral state from a nearly racemic amino acid derivative, J. American Chemical Society 130:1158–1159, 2008. Return to text.
- Viedma, C., Ortiz, J.E., de Torres, T., Izumi, T., and Blackmond, D.G., Evolution of solid phase homochirality for a proteinogenic amino acid, Evolution of solid phase homochirality for a proteinogenic amino acid, J. American Chemical Society 130:15274–15275, 2008. Return to text.
- Viedma, C., Verkuijl, B.J.V., Ortiz, J.E., de Torres, T., Kellogg, R.M. and Blackmond, D.G., Solution-Phase Racemization in the Presence of an Enantiopure Solid Phase 2010, Chemistry—a European J. 16:4932−4937, 2010. Return to text.
- Blackmond, D.G., The Origin of Biological Homochirality, Cold Spring Harb Perspectives in Biology 2:a002147, 2010. Return to text.
- Day, C., Crushing a solution of left-handed and right-handed crystals breaks its chiral symmetry, Physics Today 58(4):21–22, 2005. Return to text.
- Truman, R., Racemization of amino acids under natural conditions: part 2—kinetic and thermodynamic data, J. Creation 36(2):72–80, 2022. Return to text.
- Viedma, C., Chiral symmetry breaking during crystallization: complete chiral purity induced by nonlinear autocatalysis and recycling, Physical Review Letters 94:065504, 2005. Return to text.
- Noorduin, W.L., van Enckevort, W.J. P., Meekes, H., Kaptein, B., Kellogg, R.M., Tully, J.C., McBride, J.M., and Vlieg, E., The driving mechanism behind attrition-enhanced deracemization, Angewandte Chemie International Edition 49:8435–8438, 2010. Return to text.



Readers’ comments
Comments are automatically closed 14 days after publication.